Guided Sparse Factor Analysis Project
Yifan Zhou (zhouyf@uchicago.edu)
Introduction
Here you will find links to analysis results in the GSFA project.
A preprint of the project is available on biorxiv.
Project Background
Technologies such as CROP-seq and Perturb-seq that combine multiplexed CRISPR screening with single-cell RNA-seq (scRNA-seq) have enabled efficient readouts of transcriptome-level effects of multiple genetic perturbations in tens of thousands of individual cells in a single experiment.
Current computational approaches to detect the transcriptomic effects of perturbation:
- Differential expression analysis: assess the differential effects of perturbation one gene at a time; can be under-powered due to the sparsity and noise inherent to scRNA-seq data;
- Factor analysis followed by association of factors with perturbation: can identify "gene modules" associated with the perurbation, but subsequent analyses are necessary to interpret the biological meaning of factors, and to associate the perurbation with specific genes.
Our approach bridges factor analysis and differential expression analysis:
- Assume that the perturbation of a target gene affects certain latent factors, which in turn changes the expression of individual genes.
- Identify genetically controlled factors that are associated with the perturbation in a joint statistical framework.
- Summarize the effects of a perturbation on individual genes as the sum of effects mediated by all the factors.
GSFA Model
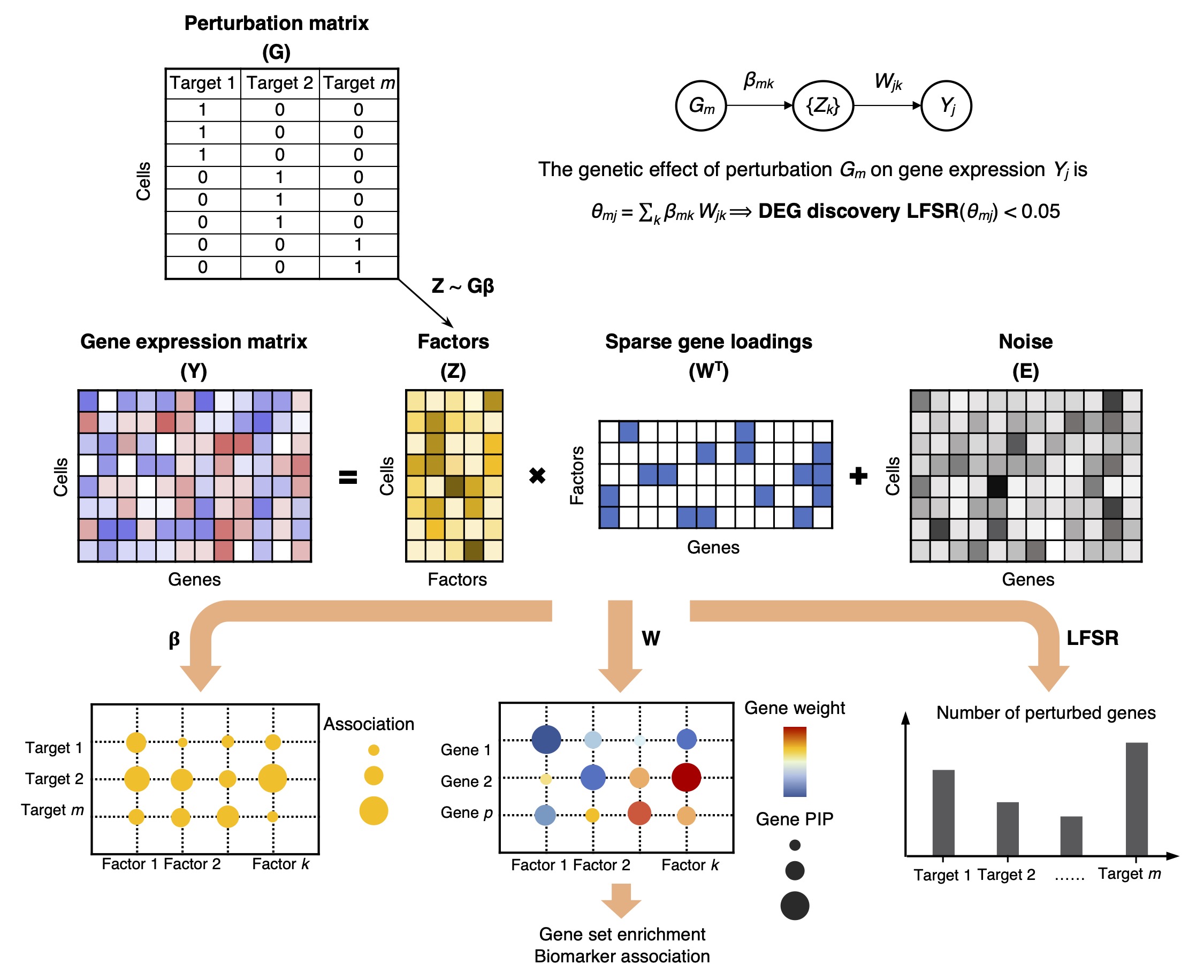
Given a matrix \(Y \in \mathbb{R}^{N \times P}\) that holds the normalizd expression levels of \(P\) genes in \(N\) samples, and a perturbation matrix \(G \in \mathbb{R}^{N \times M}\) that holds \(M\) types of sample-level perturbation conditions,
\(Y = ZW^T+E\), where \(Z \in \mathbb{R}^{N \times K}\), \(W \in \mathbb{R}^{P \times K}\), \(E_{ij} \sim N(0,\psi_j)\),
\(Z = G \beta + \Phi\), where \(\beta \in \mathbb{R}^{M \times K}\), \(\Phi_{ik} \overset{i.i.d.}{\sim} N(0,1)\).
Both \(W\) and \(\beta\) have sparse priors imposed.
Gibbs sampling is used to infer the model parameters from data.
The total effect (\(\theta_{mj}\)) of a target perturbation \(m\) on individual gene \(j\) is simply the product of the perturbation-to-factor effect and the gene-on-factor loading, summing over all \(K\) factors. The significance of this total effect is evaluated using local false sign rate (LFSR), which is similar to local false discovery rate (LFDR).
GSFA produces three main outputs: 1) the association between genetic perturbations and factors; 2) the weights of genes on factors measured by PIPs; 3) a list of DEGs of each perturbation at a given LFSR cutoff.
Simulation study
- Normal scenarios
- Count-based scenarios